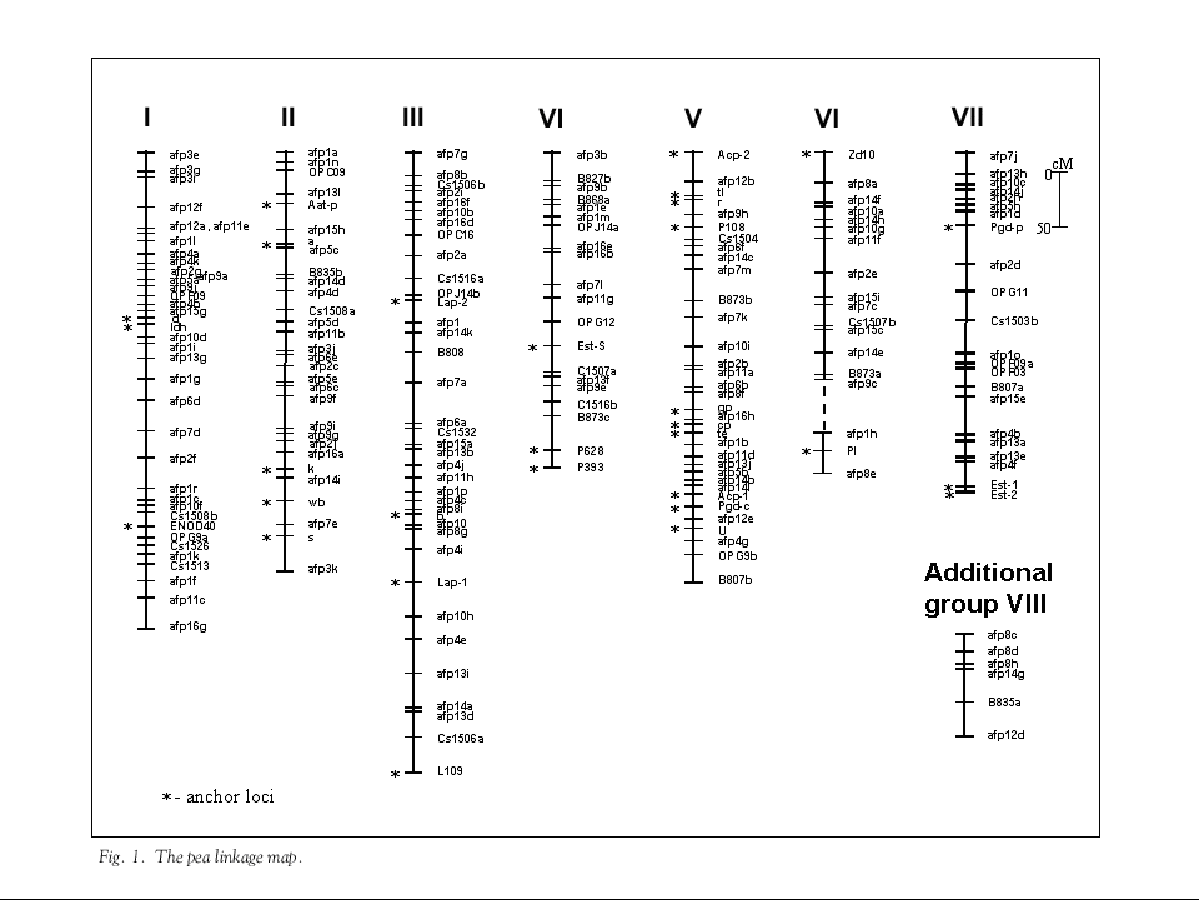
The genetic linkage map of pea (Pisum sativum L.) based on molecular, biochemical and morphological markers
Irzykowska, L., Wolko, B., and Inst. of Plant Genetics, Polish Academy of Sciences
Święcicki, W.K. Poznan, Poland
Introduction
The pea genetic map has been developing gradually during the past 50 years, with a significant spurt in the last decade. In recent years PCR-based DNA markers have been recognized to be powerful tools for the rapid construction of a genetic maps. Through the use of such a linkage map, characterization of polygenic traits can be facilitated, including identifying the genomic regions containing quantitative trait loci. In past ten years six successive, improved versions of pea map were published including the last one (14), called the consensus map.
Our map was primarily constructed with AFLP markers and supplemented with others DNA markers (RAPDs, ISSRs, CAPSs or STSs), as well as morphological and biochemical markers. Through the use of thirty anchor loci (morphological, isozyme and STS markers) the map has been related to the consensus map of the pea genome (14). The partial genetic linkage map is currently being used for quantitative trait loci (QTLs) mapping.
Materials and methods
Plant material
A total of 116 individual F2 plants from the Wt10245 x Wt11238 cross combination and F4 recombinant inbred lines (RILs) were used for map construction. Plants were evaluated in field trials. The parental lines were selected on the basis of contrasting monogenic characters as well as significant differences in quantitative trait expression. The female parent (Wt 10245) is a large seeded cultivar and the male parent (Wt11238) is a tester line with genotype that includes morphological and isozyme markers in all linkage groups.
Morphological markers
Observation of the segregation of 14 morphological monogenic characters (flower characters: A/a, B/b, K/k; leaf characters: D/d, Tl/tl, Wb/wb; pod characters: Te/te, Cp/cp, Gp/gp; seed characters: I/i, U/u, S/s, R/r, Pl/pl) were carried out on the F2 plant population.
Isozyme markers
The isozyme analysis was conducted by means of horizontal electrophoresis on 11% starch gels (3). The electrophoresis of different enzyme systems was conducted in four specific buffer systems to secure the optimal conditions of separation and stabilization of enzymes (18). The polymorphism in 18 enzyme systems was examined: aspartate aminotransferase (AAT), acid phosphatase (ACP), alcohol dehydrogenase (ADH), alanine aminotransferase (ALAT), aldolase (ALDO), diaphorase (DIA), esterase (EST), fumarase (FUM), acid b‑galactosidase (GAL 4.5), glucosephosphate isomerase (GPI), isocitrate dehydrogenase (IDH), leucine amino-peptidase (LAP), malic dehydrogenase (MDH), phosphoglucomutase (PGM), 6-phosphogluconate dehydrogenase (PGD), peroxidase (PRX) shikimate dehydrogenase (SKDH) and triosephosphate isomerase (TPI).
DNA marker analysis
Genomic DNA for PCR markers analysis was isolated from 100 mg frozen leaf tissue using Dneasy Plant mini Kit (QIAGEN).
AFLP variability analysis was conducted according to the AFLP procedure described in the AFLP analysis system I manual (GIBCO BRL). Aliquots of 250 ng DNA were digested with combination of EcoRI and MseI restriction enzymes. Selective amplification was conducted using 16 pairs of primers with three selective nucleotides. Amplification products were resolved by electrophoresis in 5% denaturing polyacrylamide gels and visualized using silver staining system (Promega).
Approximately 10 ng of genomic DNA was used as a template for RAPD amplification. 150 decameric primers obtained from Operon Technologies (Kits: OPC, OPD, OPE, OPF, OPG OPJ) and from Promega (Kits: Cs1503-1532) were used to screen the parents for polymorphism. The PCR protocol was similar to the one described by Williams et al. (17) with minor modifications.
ISSR analysis was performed using 24 primers obtained from the University of British Columbia Biotechnology Laboratory (Primer Set #9). Amplification was conducted according to the method described by Kojima et al. (5).
For STS and CAPS analysis 18 primer pairs (18-24 nucleotides) obtained from Sigma/Genosys were used. Five STS loci: Z 302, Zb 500, Zp 256 (19, 20), Zd 10 (12), Gs 185 (N.F. Weeden – pers. comm.) and 13 CAPS loci: Enod 40 (L. Wolko – pers. comm.), Q 363, L 109, P 628, P 108, P 446, I7, P 482, P 393, Q 500 (2), Hop 1 (15), Rpl 22, Pepc (16) were examined.
All RAPD, ISSR, CAPS and STS amplification products were resolved by electrophoresis in agarose gel and visualized under UV light following ethidium bromide staining.
Statistical analysis
Each marker was tested for a Mendelian segregation ratio by chi-square test using computer program Linkage 1 (10). Linkage analysis was performed with Mapmaker/EXP. 3.0 software (6). The map was created with log-likelihood score of 3.0 and map distance below 35 cM. Kosambi function was used to convert recombination values into map distances (cM)
Results
In general, the level of polymorphism in our mapping population was rather low. Fourteen genetically defined morphological and 11 isozyme polymorphisms were found. Analysis of the DNA fragment patterns generated with the sixteen AFLP primer combinations gave 166 polymorphic markers. Two of them were excluded from linkage analysis because of disturbed monogenic segregation and consequently 140 AFLP markers were mapped. The polymorphism between parental lines was screened using 150 random 10-base RAPD primers. Only 50% of them generated polymorphic bands. Many fragments were discarded due to lack of reproducibility, difficulties in scoring or incorrect segregation. Finally 33 RAPDs were used for multipoint analysis and 24 of them were mapped. Only half of the ISSR primers used generated polymorphic fragments. Ten were included in the linkage map. In the CAPS assay, 13 primer pairs were tested; each amplification product was digested with sixteen restriction enzymes to find polymorphism. Eventually, five CAPS markers were included in the linkage analysis. Five STS loci were examined, but only one was mapped. The results of chi-square test and linkage analysis were shown in Table 1. All markers used for map construction displayed expected segregation ratios.
Table 1. Results of a chi-square test and a multipoint analysis.
|
Marker type |
Monohybrid segregation |
No. analyzed markers1 |
No. linked markers |
Percentage of mapped markers |
|
AFLP |
p > 0.22 |
164 |
140 |
085.4 |
|
RAPD |
p > 0.10 |
033 |
024 |
072.7 |
|
ISSR |
p > 0.30 |
012 |
010 |
083.3 |
|
CAPS |
p > 0.47 |
005 |
005 |
100.0 |
|
STS |
p > 0.86 |
001 |
001 |
100.0 |
|
isozymes |
p > 0.35 |
011 |
011 |
100.0 |
|
morphological |
p > 0.07 |
014 |
013 |
092.9 |
1Only markers displaying expected segregation ratios were used for multipoint analysis
A genetic linkage map consisting of 204 markers (Table 1) has been developed from a mapping population of 104 RILs (F4), derived from Wt 10245 x Wt11238 cross combination. Nine linkage groups were obtained (Fig. 1). Eight of them correspond to the previously described linkage groups. The Roman numeral group designation relates to the genetic map of Weeden et al. (14). In addition one unidentified linkage group (VIII) containing AFLP and ISSR markers was found. The size of the linkage groups ranged from 34 cM to 503 cM. Thirty-six markers (15%) remained unlinked. The map spans 2416 cM with an average distance between adjacent markers of 12 cM (Kosambi units). However almost 50% of map intervals are shorter than 10 cM and only 1.5% intervals is longer than 30 cM. A brief description of each group is shown in Table 2.
Table 2. Characteristics of revealed linkage groups
|
Linkage group |
I |
II |
III |
IV |
V |
VIa |
VIb |
VII |
Unassigned |
|
No. mapped loci |
37 |
30 |
37 |
20 |
34 |
15 |
3 |
6 |
22 |
|
No. anchor loci |
3 |
5 |
4 |
3 |
10 |
1 |
1 |
3 |
0 |
|
Average distance between adjacent loci |
10 |
11 |
13 |
13 |
10 |
12 |
11 |
12 |
13 |
|
Distances shorter than 10cM (%) |
61 |
55.2 |
41.7 |
42.1 |
48.5 |
35.7 |
0 |
47.6 |
20 |
|
Length (cM) |
388 |
341 |
503 |
256 |
351 |
184 |
34 |
276 |
83 |
Linkage group I
This linkage group containing 29 AFLPs, five RAPDs, one CAPS, one isozyme and one morphological marker has been assigned to the group I of consensus map because of anchor loci d, Idh and additionally, position of Enod40 was confirmed. The distance between d and Idh is slightly shorter than in consensus map (4.5 and about 6.5 respectively). Although the morphological marker i displayed expected segregation ratios no linkages were found. More than 61% map intervals in this group were shorter than 10 cM.
Linkage group II
This linkage group, similar to the first one in length, contains five anchor markers: Aat-p and a in the upper part and k, wb, s in the lower part. The order of anchor loci was consistent with that in the consensus map. The group includes 30 markers (22 AFLP, 2 RAPD, 1 ISSR, 1 isozyme and 1 morphological). More than half the distance between adjacent loci are shorter than 10 cM.
Linkage group III
The largest linkage group (503.8 cM; 37 markers:26 AFLP, 6 RAPD, 1 CAPS, 1 ISSR, 1 morphological, 2 isozymes)) contains four markers, which were previously mapped: Lap1, Lap2, b and additionally L109. The calculated distance between b and Lap1 loci was different than that one presented in the consensus map. The average distances between adjacent loci is 13 cM.
Linkage group IV
Nineteen DNA markers (10 AFLP, 4 RAPD, 3 ISSR, 2 CAPS) and one isozyme locus were mapped in this group. The longest distance between adjacent loci was 29 cM. This group was linked to the consensus map thanks to three earlier mapped markers: EstS, P393 and P628. The linkage (14 cM) between the latter loci was confirmed.
Linkage group V
This linkage group was identified because it contained 10 anchor markers:
six morphological (r, tl, cp, gp, te and U),
three isozymes (Acp1, Acp2, Pgd-c) and one CAPS (P108). The cp-U segment
order established is somewhat different in comparison to that from consensus
map. The group includes 34 markers: 20 AFLP, 2 ISSR, 2 RAPD, 1 CAPS, 6
morphological and 3 isozymes. The average distance between adjacent loci is 10
cM.
Linkage group VI
Two groups designated as VIa and VIb were assigned to linkage group VI . Unfortunately only two markers were used as anchor loci: morphological marker Pl and STS locus Zd10. Eighteen markers were mapped altogether in this group: 14 AFLP, 1 ISSR, 1 RAPD, 1 CAPS and one morphological.
Linkage group VII
This linkage group was identified using three previously mapped isozyme loci: Pgd-p (upper part) and also the closely linked (about 4 cM) Est1 and Est2 (lower part). A total of 22 markers were included into the group: 14 AFLP, 4 RAPD, 1 ISSR and 3 isozymes. The average distance between adjacent loci is 12 cM but 48% of intervals are shorter than 10 cM.
Discussion
The relationship of the map we generated to maps published earlier was determined with the aim of increasing the utility of our map. A consensus map of the pea genome (14) consists of two parallel maps. The basic map has been developed from mapping population of RILs. The second one consists of markers, which did not segregate in the RIL population but their position was quite well documented (11). The consensus map spans about 800 cM including impressive number of mapped loci (465).
The main aim of our experiment was not creating as highly saturated a map but rather a partial map for QTL analysis. The map covers 2416 cM and comprises 204 markers distributed over nine linkage groups. The large number of unlinked markers (36) as well as about 10% map intervals longer than 25 cM reflects the need to increase markers number to cover pea genome evenly. Distances of more than 20 –25 cM between markers are common in many maps of crops (1, 2). Nevertheless the created map can be successfully used for quantitative trait loci mapping. Similar, partial maps were used for QTL mapping in cowpea (8) and faba bean (13).
The map has been developed using mainly (68.6%) AFLP markers. The AFLP technique has been reported as a reliable and reproducible assay in many crops (9) but in our analysis a medium number of loci was detected in a single assay (maximum 18 polymorphic bands).
In general, the results presented here are in good accordance with the consensus map, although there are a few differences between them. On linkage group I the position of D and Idh was confirmed. An Enod 40 marker was amplified with two specific primers constructed on the basis of cDNA nucleotide sequence obtained from the EMBL, GenBank database (Lukasz Wolko, pers. comm.). The i gene, considered as a reference marker on linkage group I did not show linkage with any other segregating marker. The yellow/green cotyledon character was described as a quantitative character by McCallum et al. (7). However, we found the 3:1 segregation ratio (p > 0.25) typical to dominant character of the i inheritance mode, but the pleiotropic effect in this trait expression might be the reason of difficulties with linkage determination.
In the case of group II, IV and VI we found the same reference marker order as in the consensus map (14). In several cases, markers segregating in our mapping population were only indirectly localized on the consensus map (e.g. a, k, P628). The postulated positions of these markers were in agreement with our data. The anchor marker order we obtained on linkage group III agrees with that suggested oin the consensus map, but the distance between B and Lap1 is larger.
Ten reference markers from the linkage group V segregated in our mapping population. Thus we could verify a marker order on a large part of this group. It was possible to confirm the localization of four markers in the upper part of the group (Acp 2, r, tl, P108). Considerable differences were found in alignment of markers in cp – U segment. Six reference markers (gp, cp, te, Acp-1, Pgd-c, U) were mapped in this part of the group. The proposed order of this marker subset is shown on Figure 1.
On the linkage group VII, the position of loci Pgd-p and Est-2 is the same as on the consensus map, but additionally the Est-1 locus was mapped. In our previous work (4), the results of linkage analysis indicated the tight linkage (about 4 cM) between Est-1 and Est-2 in each of three cross combination examined. In the present work tight linkage between esterase loci was found, so the position of Est-1 was confidently confirmed.
In summary, we found that the AFLP markers are efficient tools for quickly creating genetic map. It was possible to verify and in some cases to correct the marker localization. We hope that our results will be useful for future map updates.
1. Eujayl, I., Baum, M., Powell, W., Erskine, W. and Pehu, E. 1998. Theor. Appl. Genet. 97: 83-89.
2. Gilpin, B.J., McCallum, J.A., Frew, G.M. and Timmerman-Vaughan, G.M. 1997. Theor. Appl. Genet. 95: 1289-1299.
3. Gottlieb, L.D. 1973. Evolution, 27: 205-214.
4. Irzykowska, L., Święcicki, W.K. and Wolko, B. 2000. Vortr. Pflanzenzüchtg. 47: 98.
5. Kojima, T., Nagaoka, T., Noda, K. and Ogihara, Y. 1998. Theor. Appl. Genet. 96: 37-45.
6. Lander, E., Green, P., Abrahamson, J., Barlow, A., Daley, M., Lincoln, S. and Newburg, L. 1987. Genomics 1: 174 - 181.
7. McCallum, J. , Timmerman-Vaughan, G., Frew, T. and Russell, A. 1997. J. Amer. Hort. Sci. 122: 218-225.
8. Menendez, C.M., Hall, A.E. and Gepts, P. 1997. Theor. Appl. Genet. 95: 1210-1217.
9. Powell, W., Thomas,W.T.B., Baird, E., Lawrence, P., Booth, A., Harrower, B., McNicol, J.W., and Waugh, R. 1997. Heredity 79: 48 -59.
10. Suiter, K.A., Wendel, J.F. and Case, J. S. 1983. J. Hered. 74: 203-204.
11. Święcicki, W.K., Wolko, B. and Weeden, N.F. 2000. Vortr. Pflanzenzüchtg. 48: 65-76.
12. Timmerman, G.M., Frew, T.J., Weeden, N.F., Miller, A.L. and Goulden, D.S. 1994. Theor. Appl. Genet. 88: 1050-1055.
13. Vaz Patto, M.C., Torres, A.M., Koblizkova, A., Macas, J. and Cubero, J.I. 1999. Theor. Appl. Genet. 98: 736-743.
14. Weeden, N.F., Ellis, T.H.N., Timmerman-Vaughan, G.M., Święcicki, W.K., Rozov, S.M. and Berdnikov, V.A. 1998. Pisum Genetics 30: 1-4.
15. Weeden, N.F. and DeMason, D.A. 1999. Pisum Genetics 31: 37.
16. Weeden, N.F., Tonguc, M. and Boone, W.E. 1999. Pisum Genetics 31: 30-32.
17. Williams, J.G.K., Kubelik, A.R., Livak, K.J., Rafalski, J.A. and Tingey, S.V. 1990. Nucleic Acids Res. 18: 6531-6535.
18. Wolko, B. and Weeden, N.F. 1989. Genet. Pol. 30: 165-171.
19. Yu, J., Gu, W.K., Provvidenti, R. and Weeden, N.F. 1995. J. Amer. Soc. Hort. Sci. 120: 730-733.
20. Yu, J., Gu, W.K. and Weeden, N.F. 1996. Pisum Genetics 28: 31-32.