ITS sequence variation in selected taxa of Pisum
Saar,
D.E. and Polans, N.O.
Dept. of Biological Sciences and Plant Molecular Biology Center
Northern Illinois University, Dekalb, Illinois 60115
Most investigators currently recognize only one or two legitimate species
of Pisum (5). These usually include P.
fulvum and a P. sativum complex
comprised of two main races (humile
and elatius), weedy forms and
cultivated varieties (2). Two distinct isolates of humile
have also been described, a “northern” form that possesses the standard sativum
karyotype and a “southern” form that exhibits the same chromosomal
translocation as elatius. Despite these distinctions, there is an unmistakably close
genealogical affinity among all the wild and cultivated taxa of pea (4,5). One
approach to characterizing the nature and degree of these genetic affinities
among the various pea taxa is by comparing the nucleotide sequences of their
ribosomal DNA.
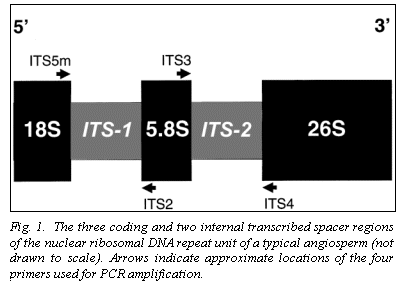 Nuclear ribosomal DNA (nrDNA) is organized as individual chromosomal
units that are repeated thousands of times in most higher plant genomes. Each of
these units contains the three genes that encode the 18S, 5.8S and 26S ribosomal
RNA subunits, as well as several different spacer DNA regions. The nucleotide
sequence variation found in both of the internal transcribed spacer regions
(ITS-1 and ITS-2, Fig. 1) is used extensively for the systematic analysis of
closely related taxa, at least in part due to the speedy rate of evolutionary
change characterizing these DNA regions (1). In this preliminary study, ITS-1
and ITS-2 DNA sequence variation is assessed for five pairs of wild and
cultivated pea taxa selected to approximate the range of Pisum. The goal of the exercise is to examine the similarity of the
sequences within paired accessions, the overall level of genetic variation found
across the entire genus, and the topological relationships established among the
five selected groups of taxa.
Nuclear ribosomal DNA (nrDNA) is organized as individual chromosomal
units that are repeated thousands of times in most higher plant genomes. Each of
these units contains the three genes that encode the 18S, 5.8S and 26S ribosomal
RNA subunits, as well as several different spacer DNA regions. The nucleotide
sequence variation found in both of the internal transcribed spacer regions
(ITS-1 and ITS-2, Fig. 1) is used extensively for the systematic analysis of
closely related taxa, at least in part due to the speedy rate of evolutionary
change characterizing these DNA regions (1). In this preliminary study, ITS-1
and ITS-2 DNA sequence variation is assessed for five pairs of wild and
cultivated pea taxa selected to approximate the range of Pisum. The goal of the exercise is to examine the similarity of the
sequences within paired accessions, the overall level of genetic variation found
across the entire genus, and the topological relationships established among the
five selected groups of taxa.
Materials and Methods
DNA for the ITS procedure is extracted from the leaves of the individual
pea plants listed in Table 1 using a CTAB protocol (7). Primers ITS2, ITS3 and
ITS4 are described elsewhere (10), as are the PCR amplification cycle and the
modified ITS5m primer (8). Gel purification (3) precedes DNA sequencing with an
Applied Biosystems model 373 DNA sequencer. PCR is performed with Perkin Elmer (Cetus)
DNA thermal cyclers. Forward and reverse DNA sequences are compared to resolve
ambiguities using PC Gene software and the resulting sequences aligned with the
Clustal X computer program. Sequence data are analyzed using the PAUP computer
package (9).
Results and Discussion
The pea ITS-1 and ITS-2 regions
examined in this study contain 298 and 349 alignable base pairs (bp),
respectively, totaling 647 bp for each of the plants analyzed. Four ambiguous
pyrimidine sites are denoted by the IUPAC/IUB
symbol “Y.” Of the 647 ITS bp sequenced for each individual plant, 629
(>97%) of these
Table
1. Variable ITS sites for 10 wild and cultivated taxa of pea.
|
|
|
Nucleotide
Position*
|
|
Taxon
|
Accession
|
ITS-1
1111111112222
0112333490346
3584259508407
|
ITS-2
11223
34080
78818
|
|
P.
fulvum Sibth.&Sm.
|
701
702
|
GTTGGGACCGATG
GTTGGGACCGATG
|
TTTAG
TTTAG
|
|
P.
sativum L. var. humile
Boiss.&Noe–(southern)
|
712
713
|
ATCAGAGCTACCA
ATCAAAGCTACCA
|
CCAAC
YCAAC
|
|
P.
sativum L. var. humile
Boiss.&Noe–(northern)
|
716
JI1794
|
GTCGGGGCTACCA
GTCGGGGCTACCA
|
CCATC
CCATC
|
|
P.
sativum L. var. elatius
Bieb.
|
721
722
|
GCCGTAGYTACCA
GCCGTAGYTACCA
|
CCATC
CCATC
|
|
P.
sativum L. cv.
‘Alaska’
‘Austrian Winter’
|
JI711
|
ACCGAAGYTACCA
ACCGAAGCTACCA
|
CCATC
CCATC
|
*In
the 5’->3’ direction (see Fig. 1), beginning with those bases nearest
primer
ITS5m
(for ITS-1) or primer ITS3 (for ITS-2). Complete sequences are available through
GenBank for ITS-1 and ITS-2, respectively, as follows: 701(AF305582, AF305920),
702(AF305583,AF305921), 712(AF305584,AF305922), 713(AF305585,AF305923),
716(AF305586,AF305924), JI1794(AF305587,AF305925), 721(AF305588,AF305926), 722
(AF305589,AF305927), Alaska(AF305202,AF305928), JI711(AF305590,AF305929).
|
|
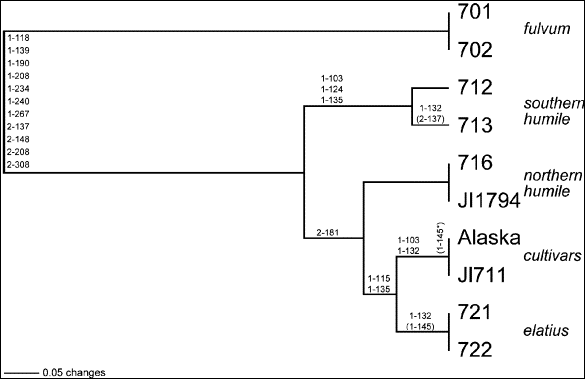
Fig.
2. UPGMA (unweighted pair
group method using arithmetic averages) phylogram of 10 wild and
cultivated pea taxa based on 18 variable ITS sites. Nucleotide
substitutions (as shown in Table 1) are located on the appropriate
branches, the first number of the designation denoting whether the variant
is derived from the ITS-1 or
ITS-2 region and the other three numbers denoting the assigned nucleotide
position within either spacer region. Parentheses indicate an ambiguous
substitution, and the asterisk indicates an ambiguous substitution within
the cv. Alaska terminus. Branch length distances are drawn with reference
to the 0.05 length standard.
|
sites are constant among the 10 pea
taxa. Only 18 of the sites are polymorphic (and only 17 are parsimony
informative). Despite its smaller size, ITS-1 contains 13 of the polymorphic
sites, as compared with the five found for ITS-2 (Table 1). These numbers attest
persuasively to both the very close evolutionary relationships that must exist
within the genus and the limited ITS information available with which to
differentiate pea taxa. By contrast, when Vicia
montbrettii (GenBank AF228075), a single taxon representing a sister genus
to Pisum, is included in the data set
for comparative purposes, more than three times as many polymorphic sites become
available.
A standard UPGMA distance analysis of the data is presented in Fig. 2. P.
fulvum, phylogenetically the most diverged from the cultivars, is assigned
as the outgroup. Actual nucleotide substitutions (24 in all) are placed on the
phylogram branches, with approximately one-half of these base changes supporting
the differentiation of fulvum from the
larger sativum ingroup. Within sativum,
all eight accessions pair according to their traditional taxonomic designations.
The selected pairs of northern humile
and elatius each displays completely
identical nucleotide sequences (at 647 sites), as does the pair of fulvum
lines comprising the outgroup. The cultivars differ at only one ambiguous site,
and the southern humile differ at only
one ambiguous and one unambiguous site. It should be noted here that accession
JI1794, listed by the John Innes Institute simply as P.
humile, seems to possess the morphological features of a northern
humile. It is thus identified in this study in accordance with its perfect
ITS sequence identity with northern humile
716.
According to the UPGMA analysis depicted in Fig. 2, elatius
is the closest taxon to the cultivated sativum,
followed by northern humile. Southern humile
is the taxon within the ingroup most distinct from the cultivars. The close
clustering of the northern and southern forms of humile
would seem intuitive, while their resolution in the phylogram supports their
established distinctiveness as well. Parsimony analyses of this same small data
set do not resolve these relationships as thoroughly as the distance model,
although they produce many of the same branches and much of the same topology.
Only the node joining the sativum
ingroup with the fulvum outgroup
receives strong (100%) support using parsimony methods. Neither branch-and-bound
nor bootstrap searches generate high clade values among the four ingroup taxa;
the single exception being a 77% bootstrap value at the node joining elatius and the cultivars.
It has been postulated that northern humile,
rather than elatius, is the closest wild progenitor of the cultivated pea, based
in part on a shared chromosomal translocation (2) and detailed chloroplast
studies (6). This compelling relationship, however, is inconsistent with the
UPGMA findings presented in Fig. 2. Northern humile is even further removed from the cultivars in a number of the
(fourteen) most parsimonious trees, in these instances reversing its position in
Fig. 2 with that presently shown for southern humile. Irrespective of the relative phylogenetic positions of
northern and southern humile, neither
this ITS data set, nor other more extensive data sets (not shown), support
northern humile as the taxon closest
to the cultivars.
Conclusions
ITS sequence variation for the selected taxa of this study suggests: 1)
very close genetic affinities throughout Pisum,
with P. fulvum exhibiting the greatest degree of divergence, 2) support
for the established taxonomic categories of the genus based upon identical or
near identical sequences within group pairs, 3) the assignment of JI1794 as a
“northern” humile, 4) the validity
of northern and southern humile as
closely-related, but distinct, lines, 5) the apparent independent evolution of a
pea chromosomal translocation and 6) a close relationship between elatius
and the cultivated sativa.
Acknowledgement: We thank Scott Grayburn for
his DNA sequencing skills. This work was supported by funds from the Department
of Biological Sciences and the Plant Molecular Biology Center, Northern Illinois
University.
1.
Baldwin, B.G., Sanderson, M.J., Porter, J.M., Wojciechowski, M.F.,
Campbell, C.S. and Donoghue, M.J. 1995. Annals
Missouri Bot. Gard. 82: 247-277.
2.
Ben Ze’ev, N. and Zohary, D. 1973.
Israel J. Bot. 22: 73-91.
3.
Dean, A.D. and Greenwald, J.E. 1995.
BioTechniques 18: 980.
4.
Hoey, B.K., Crowe, K.R., Jones, V.M. and Polans, N.O.
1996. Theor. Appl. Genet.
92: 92-100.
5.
Marx, G.A. 1977.
In Physiology of the Garden Pea. Eds. Sutcliffe, J.F. and Pate, J.S.,
Academic Press, New York, pp. 21-43.
6.
Palmer, J.D., Jorgensen, R.A. and Thompson, W.F.
1985. Genetics 109: 195-213.
7.
Saghai-Maroof, M.A., Soliman, K.M., Jorgensen, R.A. and Allard, R.W.
1984. Proc. Natl. Acad. Sci.
USA 81: 8014-8018.
8.
Sang, T., Crawford, D.J. and Stuessy, T.S.
1995. Proc. Natl. Acad. Sci. USA 92: 6813-6817.
9.
Swofford, D.L. 1998. PAUP,
Version 4.0b4a. Sinauer Associates, Sunderland,
Massachusetts.
10. White, T.J.,
Burns, T., Lee, S. and Taylor, J. 1990.
In PCR Protocols: A Guide to Methods and Applications. Eds. Innis,
M.A. and Gelfand, D.H.